Machine learning workflow enables faster, more reliable organic crystal structure prediction

Sadie Harley
scientific editor

Robert Egan
associate editor

Prediction of crystal structures of organic molecules is a critical task in many industries, especially in pharmaceuticals and design of functional materials. In pharmaceuticals, crystal structures directly influence a drug's solubility and stability.
In functional materials, like organic semiconductors, controlling crystal structures is crucial for achieving desired electronic properties. However, crystal structure prediction (CSP) is an inherently challenging task due to the weak and diverse intra- and intermolecular interactions unique to organic crystals. Even minor variations can result in entirely different packing arrangements.
CSP is typically conducted in two stages: structure exploration and structure relaxation. In the first stage, a large number of potential structures are generated, often at random, for which various search algorithms have been developed.
During structure relaxation, these structures are refined to identify the most stable configurations using energy minimization. However, random structure generation often produces several low-density and unstable structures, while conventional density functional theory (DFT)-based methods for structure relaxation are computationally expensive and time-consuming.
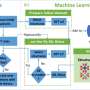
To address these challenges, Associate Professor Takuya Taniguchi from the Center for Data Science and Ryo Fukasawa from Graduate School of Advanced Science and Engineering at Waseda University, Japan, developed a breakthrough machine learning (ML)-based CSP workflow called SPaDe-CSP that leverages space group (SP) and packing density (PD) predictors.
"Our workflow employs a unique strategy where machine learning models first predict the most probable space groups and crystal densities, filtering out unstable, low-density candidates before computationally intensive relaxation steps," explains Taniguchi.
"Together with an efficient neural network potential for structure relaxation, this method enables a more direct and reliable path to identifying experimentally observed crystal arrangements."
Their study was published in the journal .
SPaDe-CSP narrows the search space for organic crystals, by first predicting probable space group candidates and crystal densities using ML models. For training and testing, the researchers extracted a dataset from the Cambridge Structural Database (CSD), consisting of 32 space group candidates with 169,656 data entries.
Both prediction models used MACCSKeys as the molecular fingerprint and LightGBM as the prediction function. The researchers also interpreted the trained models using Shapley additive explanations (SHAP) analysis to identify the most important structural characteristics for effective predictions.
After lattice sampling, the generated unrelaxed structures are then subjected to structure relaxation using an efficient neural network potential (NNP) pretrained on DFT data, ultimately producing the energy density diagram of the target molecule. Two hyperparameters control the SPaDe-CSP process: the probability threshold for filtering space groups and the tolerance window for the crystal density.
The researchers tested the workflow first on a model molecule from the CSD dataset to investigate the dependence of success rate on the hyperparameters, and then on 20 different organic molecules, including the model molecule, to test generalizability.
The results were successfully validated against the known experimental crystal structures of the molecules, and also compared against the results obtained from conventional random-CSP.
Results revealed that the probability of success increases with a higher space group threshold and a smaller density tolerance window. For 80% of the tested compounds, SPaDe-CSP successfully predicted the experimental crystal structures, achieving twice the success rate of random-CSP.
Notably, the researchers also identified a key structural descriptor correlating linearly with success rate, indicating both crystal- and molecule-level structural influences.
"Our strategy can significantly accelerate the design and discovery pipeline for new molecules within the pharmaceutical and materials science industries," says Taniguchi.
"This will enable faster, more reliable identification of the most stable, effective physical form of a new drug, important for maintaining solubility, shelf life, and overall efficacy, and allow computational screening of novel functional materials with optimal electronic properties."
By making CSP faster and more reliable, this research marks an important step toward accelerating the discovery of life-saving medication and next-generation technologies.
More information: Takuya Taniguchi et al, Crystal structure prediction of organic molecules by machine learning-based lattice sampling and structure relaxation, Digital Discovery (2025).
Provided by Waseda University